- Location : Home» Newsroom
Dynamics of duplicated gene regulatory networks governing cotton fiber development following polyploidy
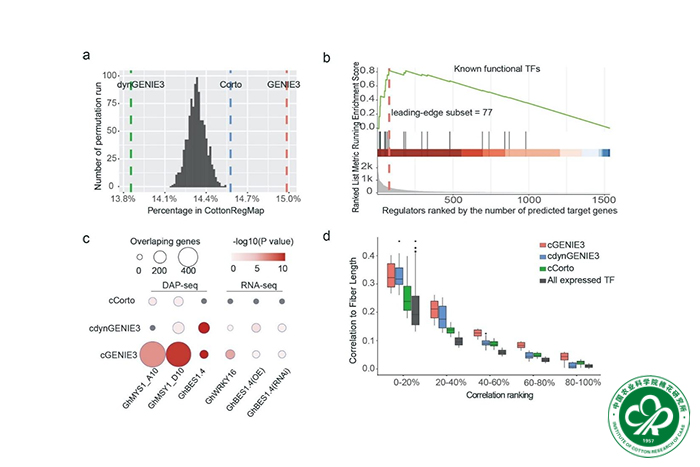
Cotton fiber development entails complex genome-wide gene regulatory networks (GRNs) that remain insufficiently resolved.
Here, we present integrative analyses of fiber GRNs using public RNA-seq datasets, integrated with genomic, transcriptomic, and cistromic data. We detail the fiber co-expression dynamics and regulatory connections, validating findings with external datasets and transcription factor (TF) binding site data.
We elucidate previously uncharacterized TFs that regulate genes involved in fiber-related functions and cellulose synthesis, and identify the regulatory role of two homoeologous G2-like TFs on fiber length. Analysis of duplicated gene expression and network relationships in allopolyploid cotton, which has two co-resident genomes (A, D), revealed novel aspects of asymmetric subgenomic developmental contributions. Whereas D-biased homoeolog pairs drive higher overall gene expression from the D subgenome, TFs from the A subgenome play a preferential regulatory role in the fiber GRN. Following allopolyploid formation, it appears that the trans-regulatory roles of TFs diversified more rapidly between homoeologs than did the cis-regulatory elements of their target genes.
Our approach underscores the utility of network analysis for detecting master regulators and provides fresh perspectives on fiber development and polyploid functional genomics through the lens of co-expression and GRN dynamics.